Let’s move on about 40 years from John
Gurdon’s work, and a decade on from Dolly. There is so much
coverage in the press about cloned mammals that we might think this
procedure has become routine and easy. The reality is that it is
still highly time-consuming and laborious to create clones by
nuclear transfer, and consequently it’s generally a very costly
process. Much of the problem lies in the fact that the process
relies on manually transferring somatic nuclei into eggs. Unlike
the amphibians that John Gurdon worked on, there’s the additional
problem that mammals don’t produce very many eggs at once.
Mammalian eggs also have to be extracted carefully from the body,
they aren’t just ejected into a tank like toad eggs. Mammalian eggs
have to be cultured incredibly delicately to keep them healthy and
alive. Researchers need to remove the nucleus manually from an egg,
inject in a nucleus from an adult cell (without damaging anything),
then keep culturing the cells really, really carefully until they
can be implanted into the uterus of another female. This is
incredibly intensive and painstaking work and we can only do it one
cell at a time.
For many years, scientists had a dream of
how they would carry out cloning in an ideal world. They would take
really accessible cells from the adult mammal they wanted to clone.
A small sample of cells scraped from the skin would be a pleasantly
easy option. Then they would treat these cells in the laboratory,
adding specific genes, or proteins, or chemicals. This treatment
would change the way the nuclei of these cells behaved. Instead of
acting like the nucleus of a skin cell, they would act the same way
as nuclei from newly fertilised eggs. The treatment would therefore
have the same ultimate effect as transferring the nuclei from adult
cells into fertilised eggs, from which their own nuclei had been
removed. The beauty of such a hypothetical scheme is that we’d have
bypassed most of the really difficult and time-consuming steps that
require such a high level of technical skill in manipulating tiny
cells. This would make it an easily accessible technique and one
that could be carried out on lots of cells simultaneously, rather
than just one nuclear transfer at a time.
Okay, we’d still have to find a way of
putting them into a surrogate mother, but we only have to go down
the surrogate mother route if we want to generate a complete
individual. Sometimes this is exactly what we want – to re-create a
prize bull or prize stallion, for example, but this is not what
most sane people want to do with humans. Indeed cloning humans
(reproductive cloning) is banned in pretty much every country which
has the scientists and the infrastructure to undertake such a task.
But actually for most purposes we don’t need to go as far as this
stage for cloning to be useful for humans. What we need are cells
that have the potential to turn into lots of other cell types.
These are the cells that are known as stem cells, and they are
metaphorically near the top of Waddington’s epigenetic landscape.
The reason we need such cells lies in the nature of the diseases
that are major problems in the developed world.
In the rich parts of our planet the
diseases that kill most of us are chronic. They take a long time to
develop and often they take a long time to kill us when they do.
Take heart disease, for example – if someone survives the initial
heart attack they don’t necessarily ever go back to having a
totally healthy heart again. During the attack some of the heart
muscle cells (cardiomyocytes) may become starved of oxygen and die.
We might imagine this would be no problem, as surely the heart can
create replacement cells? After all, if we donate blood, our bone
marrow can make more red blood cells. Similarly, we have to do an
awful lot of damage to the liver before it stops being able to
regenerate and repair itself. But the heart is different.
Cardiomyocytes are referred to as ‘terminally differentiated’ –
they have gone right to the bottom of Waddington’s hill and are
stuck in a particular trough. Unlike bone marrow or liver, the
heart doesn’t have an accessible reservoir of less specialised
cells (cardiac stem cells) that could turn into new cardiomyocytes.
So, the long-term problem that follows a heart attack is that our
bodies can’t make new cardiac muscle cells. The body does the only
thing it can and replaces the dead cardiomyocytes with connective
tissue, and the heart never beats in quite the same way it did
before.
Similar things happen in so many diseases
– the insulin-secreting cells that are lost when teenagers develop
type 1 diabetes, the brain cells that are lost in Alzheimer’s
disease, the cartilage producing cells that disappear during
osteoarthritis – the list goes on and on. It would be great if we
could replace these with new cells, identical to our own. This way
we wouldn’t have to deal with all the rejection issues that make
organ transplants such a challenge, or with the lack of
availability of donors. Using stem cells in this way is referred to
as therapeutic cloning; creating cells identical to a specific
individual in order to treat a disease.
For over 40 years we’ve known that in
theory this could be possible. John Gurdon’s work and all that
followed after him showed that adult cells contain the blueprints
for all the cells of the body if we can only find the correct way
of accessing them. John Gurdon had taken nuclei from adult toads,
put them into toad eggs and been able to push those nuclei all the
way back up Waddington’s landscape and create new animals. The
adult nuclei had been – and this word is critical – reprogrammed.
Ian Wilmut and Keith Campbell had done pretty much the same thing
with sheep. The important common feature to recognise here is that
in each case the reprogramming only worked when the adult nucleus
was placed inside an unfertilised egg. It was the egg that was
really important. We can’t clone an animal by taking an adult
nucleus and putting it into some other cell type.
Why not?
We need a little cell biology here. The
nucleus contains the vast majority of the DNA/genes that encode us
– our blueprint. There’s a miniscule fraction of DNA that isn’t in
the nucleus, it’s in tiny structures called mitochondria, but we
don’t need to worry about that here. When we’re first taught about
cells in school it’s almost as if the nucleus is all powerful and
the rest of the cell – the cytoplasm – is a bag of liquid that
doesn’t really do much. Nothing could be further from the truth,
and this is especially the case for the egg, because the toads and
Dolly have taught us that the cytoplasm of the egg is absolutely
key. Something, or some things, in that egg cytoplasm actively
reprogrammed the adult nucleus that the experimenters injected into
it. These unknown factors moved a nucleus from the bottom of one of
Waddington’s troughs right back to the top of the
landscape.
Nobody really understood how the
cytoplasm of eggs could convert adult nuclei into ones like
zygotes. There was pretty much an assumption that whatever it was
must be incredibly complicated and difficult to unravel. Often in
science really big questions have smaller, more manageable
questions inside them. So a number of labs tackled a conceptually
simpler, but technically still hugely challenging
issue.
Endless potential
Remember that ball at the top of
Waddington’s landscape. In cellular terms it’s the zygote and it’s
referred to as totipotent, that is, it has the potential to form
every cell in the body, including the placenta. Of course, zygotes
by definition are rather limited in number and most scientists
working in very early development use cells from a bit later, the
famous embryonic stem (ES) cells. These are created as a result of
normal developmental pathways. The zygote divides a few times to
create a bundle of cells called the blastocyst. Although the
blastocyst typically has less than 150 cells it’s already an early
embryo with two distinct compartments. There’s an outer layer
called the trophectoderm, which will eventually form the placenta
and other extra-embryonic tissues, and an inner cell mass
(ICM).
Figure 2.1 shows
what the blastocyst looks like. The drawing is in two dimensions
but in reality the blastocyst is a three-dimensional structure, so
the actual shape is that of a tennis ball that’s had a golf ball
glued inside it.
The cells of the ICM can be grown in the
lab in culture dishes. They’re fiddly to maintain and require
specialised culture conditions and careful handling, but do it
right and they reward us by dividing a limitless number of times
and staying the same as the parent cell. These are the ES cells and
as their full name suggests, they can form every cell of the embryo
and ultimately of the mature animal. They aren’t totipotent – they
can’t make placenta – so they are called pluripotent because they
make pretty much anything else.
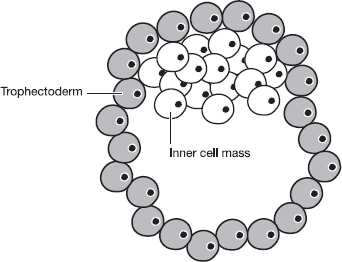
Figure 2.1 A diagram
of the mammalian blastocyst. The cells of the trophectoderm will
give rise to the placenta. During normal development, the cells of
the Inner Cell Mass (ICM) will give rise to the tissues of the
embryo. Under laboratory conditions, the cells of the ICM can be
grown in culture as pluripotent embryonic stem (ES)
cells.
These ES cells have been invaluable for
understanding what’s important for keeping cells in a pluripotent
state. Over the years a number of leading scientists including Azim
Surani in Cambridge, Austin Smith in Edinburgh, Rudolf Jaenisch in
Boston and Shinya Yamanaka in Kyoto have devoted huge amounts of
time to identifying the genes and proteins expressed (switched on)
in ES cells. They particularly tried to identify genes that keep
the ES cells in a pluripotent state. These genes are
extraordinarily important because ES cells seem to be very prone to
turn into other cell types in culture if you don’t keep the
conditions just right. Just a small change in culture conditions,
for example, and a culture dish full of one-time ES cells can
differentiate into cardiomyocytes and do what heart cells do best:
they beat along in time with one another. A slightly different
change in conditions – altering the delicate balance of chemicals
in the culture fluid, for example, can divert the ES cells away
from the cardiac route and start the development of cells that give
rise to the neurons in our brains.
Scientists working on ES cells identified
a whole slew of genes that were important for keeping the cells
pluripotent. The functions of the various genes they identified
weren’t necessarily identical. Some were important for
self-renewal, i.e. one ES dividing to form two ES cells, whereas
others were required to stop the cells from differentiating1.
So, by the early years of the 21st
century scientists had found a way of maintaining pluripotent ES
cells in culture dishes and they knew quite a lot about their
biology. They had also worked out how to change the culture
conditions so that the ES cells would differentiate into various
cell types including liver cells, heart cells, neurons etc. But how
does this help with the dream we laid out earlier? Could the labs
use this information to create new ways of driving cells backwards,
to the top of Waddington’s landscape? Would it be possible to take
a fully differentiated cell and treat it in a lab so that it would
become just like an ES cell, with all the potential that implies?
Whilst scientists had good reason to believe this would be
theoretically possible, that’s a long way from actually being able
to do it. But it was a wonderfully tantalising prospect for
scientists interested in using stem cells to treat human
diseases.
By the middle of the first decade of this
century, over twenty genes had been identified that seemed to be
critical to ES cells. It wasn’t necessarily clear how they worked
together and there was every reason to think that there was still
plenty we didn’t understand about the biology of ES cells. It was
assumed that it would be almost inconceivably difficult to take a
mature cell and essentially recreate the vastly complex
intracellular conditions that are found in an ES cell.
The triumph of optimism
Sometimes the greatest scientific
breakthroughs happen because someone ignores the prevailing
pessimism. In this case, the optimist who decided to test what
everyone else had assumed was impossible was the aforementioned
Shinya Yamanaka, with his postdoctoral research associate Kazutoshi
Takahashi.
Professor Yamanaka is one of the youngest
luminaries in the stem cell and pluripotency field. He was born in
Osaka in the early 1960s and rather unusually he has held
successful academic positions in high profile institutions in both
Japan and the USA. He originally trained as a clinician and became
an orthopaedic surgeon. Specialists in this discipline are
sometimes dismissed by other surgeons as ‘the hammer and chisel
brigade’. This is unfair, but it is true that orthopaedic surgical
practice is about as far away from elegant molecular biology and
stem cell science as it’s possible to get.
Perhaps more than any of the other
researchers working in the stem cell field, Professor Yamanaka had
been driven by a desire to find a way of creating pluripotent cells
from differentiated cells in a lab. He started this stage of his
work with a list of 24 genes which were vitally important in ES
cells. These were all genes called ‘pluripotency genes’ – they have
to be switched on if ES cells are to remain pluripotent. If you use
various experimental techniques to switch these genes off, the ES
cells start to differentiate, just like those beating heart cells
in the culture dish, and they never revert to being ES cells again.
Indeed, that is partly what happens quite naturally during
mammalian development, when cells differentiate and become
specialised – they switch off these pluripotency
genes.
Shinya Yamanaka decided to test if
combinations of these genes would drive differentiated cells
backwards to a more primitive developmental stage. It seemed a long
shot and there was always the worry that if the results were
negative – i.e. if none of the cells went ‘backwards’ – he wouldn’t
know if it was because it just wasn’t possible or if he just hadn’t
got the experimental conditions right. This was a risk for an
established scientist like Yamanaka, but it was an even bigger
gamble for a relatively junior associate like Takahashi, because of
the way that the scientific career ladder works.
When faced with the exposure of damaging
personal love letters, the Duke of Wellington famously responded,
‘Publish and be damned!’ The mantra for scientists is almost the
same but differs in one critical respect. For us, it’s ‘publish
or be damned’ – if you don’t publish papers, you can’t get
research funding and you can’t get jobs in universities. And it is
rare indeed to get a paper into a good journal if the message of
your years of effort boils down to, ‘I tried and I tried but it
didn’t work.’ So to take on a project with relatively little
likelihood of positive results is a huge leap of faith and we have
to admire Takahashi’s courage, in particular.
Yamanaka and Takahashi chose their 24
genes and decided
to test them in a cell type known as MEFs
– mouse embryonic fibroblasts. Fibroblasts are the main cells in
connective tissue and are found in all sorts of organs including
skin. They’re really easy to extract and they grow very easily in
culture, so are a great source of cells for experiments. Because
the ones known as MEFs are from embryos the hope was that they
would still retain a bit of capacity to revert to very early cell
types under the right conditions.
Remember how John Gurdon used donor and
acceptor toad strains that had different genetically-encoded
markers, so he could tell which nuclei had generated the new
animals? Yamanaka did something similar. He used cells from mice
which had an extra gene added. This gene is called the neomycin
resistance (neoR) gene and it does
exactly what it says on the can. Neomycin is an antibiotic-type
compound that normally kills mammalian cells. But if the cells have
been genetically engineered to express the
neoR gene, they will survive. When
Yamanaka created the mice he needed for his experiments he inserted
the neoR gene in a particular way.
This meant that the neoR gene
would only get switched on if the cell it was in had become
pluripotent. The cell had to be behaving like an ES cell. So if his
experiments to push the fibroblasts backwards experimentally into
the undifferentiated ES cell state were successful, the cells would
keep growing, even when a lethal dose of the antibiotic was added.
If the experiments were unsuccessful, all the cells would
die.
Professor Yamanaka and Doctor Takahashi
inserted the 24 genes they wanted to test into specially designed
molecules called vectors. These act like Trojan horses, carrying
high concentrations of the ‘extra’ DNA into the fibroblasts. Once
in the cell, the genes were switched on and produced their specific
proteins. Introducing these vectors can be done relatively easily
on a large number of cells at once, using chemical treatments or
electrical pulses (no fiddly micro-injections for Yamanaka, no
indeed). When Shinya Yamanaka used all 24 genes simultaneously,
some of the cells survived the neomycin treatment. It was only a
tiny fraction of the cells but it was an encouraging result
nonetheless. It meant these cells had switched on the
neoR gene. This implied they were
behaving like ES cells. But if he used the genes singly, no cells
survived. Shinya Yamanaka and Kazutoshi Takahashi then added
various sets of 23 genes to the cells. They used the results from
these experiments to identify ten genes that were each really
critical for creating the neomycin-resistant pluripotent cells. By
testing various combinations from these ten genes they finally hit
on the smallest number of genes that could act together to turn
embryonic fibroblasts into ES-like cells.
The magic number turned out to be four.
When the fibroblasts were invaded by vectors carrying genes called
Oct4, Sox2, Klf4 and c-Myc something
quite extraordinary happened. The cells survived in neomycin,
showing they had switched on the
neoR gene and were therefore like
ES cells. Not only that, but the fibroblasts began to change shape
to look like ES cells. Using various experimental systems, the
researchers were able to turn these reprogrammed cells into the
three major tissue types from which all organs of the mammalian
body are formed – ectoderm, mesoderm and endoderm. Normal ES cells
can also do this. Fibroblasts never can. Shinya Yamanaka then
showed that he could repeat the whole process using fibroblasts
from adult mice rather than embryos as his starting material. This
showed that his method didn’t rely on some special feature of
embryonic cells, but could also be applied to cells from completely
differentiated and mature organisms.
Yamanaka called the cells that he created
‘induced pluripotent stem cells’ and the acronym – iPS cells – is
now familiar terminology to everyone working in biology. When we
consider that this phrase didn’t even exist five years ago, its
universal recognition amongst scientists shows just how important a
breakthrough this really is.
It’s incredible to think that mammalian
cells carry about 20,000 genes, and yet it only takes four to turn
a fully differentiated cell into something that is pluripotent.
With just four genes Professor Yamanaka was able to push the ball
right from the bottom of one of Waddington’s troughs, all the way
back up to the top of the landscape.
It wasn’t surprising that Shinya Yamanaka
and Kazutoshi Takahashi published their findings in Cell,
the world’s most prestigious biological journal2. What
was a bit surprising was the reaction. Everyone in 2006 knew this
was huge, but they knew it was only huge if it was right. An awful
lot of scientists couldn’t really believe that it was. They didn’t
for one moment think that Professor Yamanaka and Doctor Takahashi
were lying, or had done anything fraudulent. They just thought they
had probably got something wrong, because really, it couldn’t be
that simple. It was analogous to someone searching for the Holy
Grail and finding it the second place they looked, under the peas
at the back of the freezer.
The obvious thing of course would be for
someone to repeat Yamanaka’s work and see if they could get the
same results. It may seem odd to people working outside science,
but there wasn’t an avalanche of labs that wanted to do this. It
had taken Shinya Yamanaka and Kazutoshi Takahashi two years to run
their experiments, which were time-consuming and required
meticulous control of all stages. Labs would also be heavily
committed to their existing programmes of research and didn’t
necessarily want to be diverted. Additionally, the organisations
that fund researchers to carry out specific programmes of work are
apt to look a bit askance if a lab head suddenly abandons a
programme of agreed research to do something entirely different.
This would be particularly damaging if the end result was a load of
negative data. Effectively, that meant that only an exceptionally
well-funded lab, with the best equipment and a very self-confident
head, would even think of ‘wasting time’ repeating someone else’s
experiments.
Rudolf Jaenisch from The Whitehead
Institute in Cambridge, MA is a colossus in the field of creating
genetically engineered animals. Originally from Germany, he has
worked in the USA for almost the last 30 years. With curly grey
hair and a frankly impressive moustache, he is immediately
recognisable at conferences. It was perhaps unsurprising that he
was the scientist who took the risk of diverting some of the work
in his lab to see if Shinya Yamanaka really had achieved the
seemingly impossible. After all, Rudolf Jaenisch is on record
stating that, ‘I have done many high risk projects through the
years, but I believe that if you have an exciting idea, you must
live with the chance of failure and pursue the
experiment.’
At a conference in Colorado in April 2007
Professor Jaenisch stood up to give his presentation and announced
that he had repeated Yamanaka’s experiments. They worked. Yamanaka
was right. You could make iPS cells by introducing just four genes
into a differentiated cell. The effect on the audience was
dramatic. The atmosphere was like one of those great moments in old
movies where the jury delivers its verdict and all the hacks dash
off to call the editor.
Rudolf Jaenisch was gracious – he freely
conceded that he had carried out the experiments because he just
knew that Yamanaka couldn’t be right. The field went crazy after
that. First, the really big labs involved in stem cell research
started using Yamanaka’s technique, refining and improving it so it
worked more efficiently. Within a couple of years even labs that
had never cultured a single ES cell were generating iPS cells from
tissues and donors they were interested in. Papers on iPS cells are
now published every week of the year. The technique has been
adapted for direct conversion of human fibroblasts into human
neuronal cells without having to create iPS cells first3. This
is equivalent to rolling a ball halfway up Waddington’s epigenetic
landscape and then back down into a different trough.
It’s hard not to wonder if it was
frustrating for Shinya Yamanaka that nobody else seemed to take up
his work until the American laboratory showed that he was right. He
shared the 2009 Lasker Prize with John Gurdon so maybe he’s not
really all that concerned. His reputation is now
assured.
Follow the money
If all we read is the scientific
literature, then the narrative for this story is quite inspiring
and fairly straightforward. But there’s another source of
information, and that’s the patent landscape, which typically
doesn’t emerge from the mist until some time after the papers in
the peer-reviewed journals. Once the patent applications in this
field started appearing, a somewhat more complicated tale began to
unfold. It takes a while for this to happen, because patents remain
confidential for the first year to eighteen months after they are
submitted to the patent offices. This is to protect the interests
of the inventors, as this period of grace gives them time to get on
with work on confidential areas without declaring to the world what
they’ve invented. The important thing to realise is that both
Yamanaka and Jaenisch have filed patents on their research into
controlling cell fate. Both of these patent applications have been
granted and it is likely that cases will go to court to test who
can really get protection for what. And the odd thing, given that
Yamanaka published first, is the fact that Jaenisch filed a
patent on this field before him.
How could that be? It’s partly because a
patent application can be quite speculative. The applicant doesn’t
have to have proof of every single thing that they claim. They can
use the grace period to try to obtain some proof to support their
assertions from the original claim. In US legal terms Shinya
Yamanaka’s patent dates from 13 December 2005 and covers the work
described a few paragraphs ago – how to take a somatic cell and use
the four factors – Oct4, Sox2, Klf4 and
c-Myc – to turn it into a pluripotent cell. Rudolf
Jaenisch’s patent potentially could have a legal first date of 26
November 2003. It contains a number of technical aspects and it
makes claims around expressing a pluripotency gene in a somatic
cell. One of the genes it suggests is Oct4. Oct4 had been
known for some time to be vital for the pluripotent state, after
all, that’s one of the reasons why Yamanaka had included it in his
original reprogramming experiments. The legal arguments around
these patents are likely to run and run.
But why did these labs, run by fabulous
and highly creative scientists, file these patents in the first
place? Theoretically, a patent allows the holder access to an
exclusive means of doing something. However, in academic circles
nobody ever tries to stop an academic scientist in another lab from
running a basic science experiment. What the patent is really for
is to make sure that the original inventor makes money out of their
good idea, instead of other people cashing in on their
inventiveness.
The most profitable patents of all in
biology tend to be things that can be used to treat disease in
people, or that help researchers to develop new treatments faster.
And that’s why there is going to be such a battle over the Jaenisch
and Yamanaka patents. The courts may decide that every time someone
makes iPS cells, money will have to be paid to the researchers and
institutions who own the original ideas. If companies sell iPS
cells that they make, and have to give a percentage of the income
back to the patent holders, the potential returns could be
substantial. It’s worth looking at why these cells are viewed as
potentially so valuable in monetary terms.
Let’s take just one disease, type 1
diabetes. This typically starts in childhood when certain cells in
the pancreas (the delightfully named beta cells in the Islets of
Langerhans) are destroyed through processes that aren’t yet clear.
Once lost, these cells never grow back and as a consequence the
patient is no longer able to produce the hormone insulin. Without
insulin it’s impossible to control blood sugar levels and the
consequences of this are potentially catastrophic. Until we found
ways of extracting insulin from pigs and administering it to
patients, children and young adults routinely died as a result of
diabetes. Even now, when we can administer insulin relatively
easily (normally an artificially synthesised human form), there are
a lot of drawbacks. Patients have to monitor their blood sugar
levels multiple times a day and alter their insulin dose and food
intake to try and stay within certain boundaries. It’s hard to do
this consistently over many years, especially for a teenager. How
many adolescents are motivated by things that might go wrong when
they are 40? Long-term type 1 diabetics are prone to a vast range
of complications, including loss of vision, poor circulation that
can lead to amputations, and kidney disease.
It would be great if, instead of
injecting insulin every day, diabetics could just receive new beta
cells. The patient could then produce their own insulin once more.
The body’s own internal mechanisms are usually really good at
controlling blood sugar levels so most of the complications would
probably be avoided. The problem is that there are no cells in the
body that are able to create beta cells (they are at the bottom of
one of Waddington’s troughs) so we would need to use either a
pancreas transplant or perhaps change some human ES cells into beta
cells and put those into the patient.
There are two big problems in doing this.
The first is that donor materials (either ES cells or a whole
pancreas) are in short supply so there’s nowhere near enough to
supply all the diabetics. But even if there were enough, there’s
still the problem that they won’t be the same as the patient’s
tissues. The patient’s immune system will recognise them as foreign
and try to reject them. The person might be able to come off
insulin but would probably need to be on immuno-suppressive drugs
all their life. This is not really that much of a trade-off, as
these drugs have a range of pretty awful side-effects.
iPS cells suddenly create a new way
forwards. Take a small scraping of skin cells from our patient,
whom we shall call Freddy. Grow these cells in culture until we
have enough to work with (this is pretty easy). Use the four
Yamanaka factors to create a large number of iPS cells, treat these
in the lab to turn them into beta cells and put them back into the
patient. There will be no immune rejection because Freddy will just
be receiving Freddy cells. Recently, researchers have shown they
can do exactly this in mouse models of diabetes4.
It won’t be that simple of course. There
are a whole range of technological hurdles to overcome, not least
the fact that one of the four Yamanaka factors, c-Myc, is
known to promote cancer. But in the few years since that key
publication in Cell, substantial progress has been made in
improving the technology so that it is moving ever closer to the
clinic. It’s possible to make human iPS cells pretty much as easily
as mouse ones and you don’t always need to use c-Myc5.
There are ways of creating the cells that take away some of the
other worrying safety problems as well. For example, the first
methods for creating iPS cells used animal products in the cell
culture stages. This is always a worry, because of fears about
transmitting weird animal diseases into the human population. But
researchers have now found synthetic replacements for these animal
products6. The whole field of iPS
production is getting better all the time. But we’re not over the
line yet.
One of the problems commercially is that
we don’t yet know what the regulatory authorities will demand by
way of safety and supporting data before they let iPS cells be used
in humans. Currently, licensing iPS cells for therapeutic use would
involve two different areas of medical regulation. This is because
we would be giving a patient cells (cell therapy) which had been
genetically modified (gene therapy). Regulators are wary
particularly because so many of the gene therapy trials that were
launched with such enthusiasm in the 1980s and 1990s either had
little benefit for the patient or sometimes even terrible and
unforeseen consequences, including induction of lethal
cancers7. The number of potentially
costly regulatory hurdles iPS cells will have to get over before
they can be given to patients is huge. We might think no investor
would put any money into something so potentially risky. Yet invest
they do, and that’s because if researchers can get this technology
right the return on the investment could be huge.
Here’s just one calculation. At a
conservative estimate, it costs about $500 per month in the United
States to supply insulin and blood sugar monitoring equipment for a
diabetic. That’s $6,000 a year, so if a patient lives with diabetes
for 40 years that’s $240,000 over their lifetime. Then add in the
costs of all the treatments that even well-managed diabetic
patients will need for the complications they are likely to suffer
because of their illness. It’s fairly easy to see how each
patient’s diabetes-related lifetime healthcare costs could be at
least a million dollars. And there are at least a million type 1
diabetics in the US alone. This means that at the very least, the
US economy spends over a billion dollars every four years, just in
treating type 1 diabetes. So even if iPS cells cost a lot to get
into the clinic, they have the potential to make an enormous return
on investment if they work out cheaper than the lifetime cost of
current therapies.
That’s just for diabetes. There are a
whole host of other diseases for which iPS cells could provide an
answer. Just a few examples include patients with blood clotting
disorders, such as haemophilias; Parkinson’s disease;
osteo-arthritis and blindness caused by macular degeneration. As
science and technology get better at creating artificial structures
that can be implanted into our bodies, iPS cells will be used for
replacing damaged blood vessels in heart disease, and regenerating
tissues destroyed by cancer or its treatment.
The US Department of Defense is providing
funding into iPS cells. The military always needs plenty of blood
in any combat situation so that it can treat wounded personnel. Red
blood cells aren’t like most cells in our bodies. They have no
nucleus, which means they can’t divide to form new cells. This
makes red blood cells a relatively safe type of iPS cell to start
using clinically, as they won’t stay in the body for more than a
few weeks. We also don’t reject these cells in the same way that we
would a donor kidney, for example, because there are differences in
the ways our immune systems recognise these cells. Different people
can have compatible red blood cells – it’s the famous ABO blood
type system, plus some added complications. It’s been calculated
that we could take just 40 donors of specific blood types, and
create a bank of iPS cells from those people that would supply all
our needs8. Because iPS cells can keep
on dividing to create more iPS cells when grown under the right
conditions, we could create a never-ending bank of cells. There are
well-established methods for taking immature blood stem cells and
growing them under specific stimuli so that they will differentiate
to form (ultimately) red blood cells. Essentially, it should be
possible to create a huge bank of different types of red blood
cells, so that we can always have matching blood for patients, be
these from the battlefield or a traffic accident.
The generation of iPS cells has been one
of those rare events in biology that have not just changed a field,
but have almost reinvented it. Shinya Yamanaka is considered by
most to be a dead cert to share a Nobel Prize with John Gurdon in
the near future, and it would be difficult to over-estimate the
technological impact of the work. But even though the achievement
is extraordinary, nature already does so much more, so much
faster.
When a sperm and an egg fuse, the two
nuclei are reprogrammed by the cytoplasm of the egg. The sperm
nucleus, in particular, very quickly loses most of the molecular
memory of what it was and becomes an almost blank canvas. It’s this
reprogramming phenomenon that was exploited by John Gurdon, and by
Ian Wilmut and Keith Campbell, when they inserted adult nuclei into
the cytoplasm of eggs and created new clones.
When an egg and sperm fuse, the
reprogramming process is incredibly efficient and is all over
within 36 hours. When Shinya Yamanaka first created iPS cells only
a miniscule number, a fraction far less than 1 per cent of the
cells in the best experiment, were reprogrammed. It literally took
weeks for the first reprogrammed iPS cells to grow. A lot of
progress has been made in improving the percentage efficiency and
speed of reprogramming adult cells into iPS cells, but it still
doesn’t come within spitting range of what happens during normal
fertilisation. Why not?
The answer is epigenetics. Differentiated
cells are epigenetically modified in specific ways, at a molecular
level. This is why skin fibroblasts will normally always remain as
skin fibroblasts and not turn into cardiomyocytes, for example.
When differentiated cells are reprogrammed to become pluripotent
cells – whether by somatic cell nuclear transfer or by the use of
the four Yamanaka factors – the differentiation-specific epigenetic
signature must be removed so that the nucleus becomes more like
that of a newly fertilised zygote.
The cytoplasm of an egg is incredibly
efficient at reversing the epigenetic memory on our genes, acting
as a giant molecular eraser. This is what it does very rapidly when
the egg and sperm nuclei fuse to form a zygote. Artificial
reprogramming to create iPS cells is more like watching a
six-year-old doing their homework – they are forever rubbing out
the wrong bit whilst leaving in the mis-spelt words, and then
tearing a hole in the page because they rub too vigorously.
Although we are starting to get a handle on some of the processes
involved, we are a long way from recreating in the lab what happens
naturally.
Until now we have been talking about
epigenetics at the phenomenon scale. The time has come to move into
the molecules that underlie all the remarkable events we’ve talked
about so far, and many more besides.