At a purely biological, and especially
an anatomical level, men and women are different. There are ongoing
debates about whether or not certain behaviours, ranging from
aggression to spatial processing, have a biological gender bias.
But there are certain physical characteristics that are linked
unequivocally to gender. One of the most fundamental differences is
in the reproductive organs. Women have ovaries, men have testicles.
Women have a vagina and a uterus, men have a penis.
There is a clear biological basis to
this, and perhaps unsurprisingly, it’s all down to genes and
chromosomes. Humans have 23 pairs of chromosomes in their cells,
and inherited one of each pair from each parent. Twenty-two of
these pairs (imaginatively named chromosomes 1 to 22) are called
autosomes and each member of a specific pair of autosomes looks
very similar. By ‘looks’ we mean exactly that. At a certain stage
in cell division the DNA in chromosomes becomes exceptionally
tightly coiled up. If we use the right techniques we can actually
see chromosomes down a microscope. These chromosomes can be
photographed. In pre-digital days, clinical geneticists literally
used to cut out the pictures of the individual chromosomes with a
pair of scissors and rearrange them in pairs to create a nice
orderly picture. These days the image processing can be carried out
by a computer, but in either case the result is a picture of all
the chromosomes in a cell. This picture is called a
karyotype.
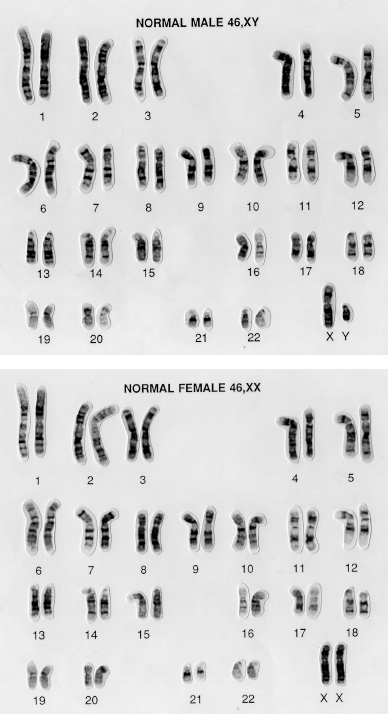
Figure 9.1 Karyotype
of all the chromosomes in a male (top) and female (bottom) somatic
cell. Note that the female cell contains two X chromosomes and no Y
chromosome; the male cell contains one X chromosome and one Y
chromosome. Note also the substantial difference in size between
the X and Y chromosomes. Photos: Wessex Reg Genetics
Centre/Wellcome Images.
Karyotype analysis is how scientists
originally discovered that there were three copies of chromosome 21
in the cells of people with Down’s syndrome. This is known as
trisomy 21.
When we produce a human karyotype from a
female, there are 23 pairs of identical chromosomes. But if we
create a human karyotype from a male, the picture is different, as
we can see in Figure 9.1. There are 22
obvious pairs – the autosomes – but there are two chromosomes left
over that don’t look like each other at all. One is very large, one
exceptionally small. These are called the sex chromosomes. The
large one is called X, and the small one is called Y. The notation
to describe the normal chromosome constitution of human males is
46, XY. Females are described as 46, XX because they don’t have a Y
chromosome, and instead have two X chromosomes.
The Y chromosome carries very few active
genes. There are only between 40 and 50 protein-coding genes on the
Y chromosome, of which about half are completely male-specific. The
male-specific genes only occur on the Y chromosome, so females have
no copies of these. Many of these genes are required for
male-specific aspects of reproduction. The most important one in
terms of sex determination is a gene called SRY. SRY
proteins activate a testis-determining pathway in the embryo. This
leads to production of testosterone, the archetypal ‘male’ hormone,
which then masculinises the embryo.
Occasionally, individuals who
phenotypically appear to be girls have the male 46, XY karyotype.
In these cases the SRY gene is often inactive or deleted and
consequently the foetus develops down the default female
pathway1. Sometimes, the other
scenario arises. Individuals who phenotypically appear to be boys
can have the typically female karyotype of 46, XX. In these cases a
tiny section of the Y chromosome containing the SRY gene has
often transferred onto another chromosome during formation of sperm
in the father. This is enough to drive masculinisation of the
foetus2. The region of the Y
chromosome that was transferred was too small to be detected by the
karyotyping process.
The X chromosome is very different. The X
chromosome is extremely large and carries about 1300 genes. A
disproportionate number of these genes are involved in brain
function. Many are also required for various stages in formation of
the ovaries or the testes, and for other aspects of fertility in
both males and females3.
Getting the dose right
So, about 1300 genes on the X
chromosome. That creates an interesting problem. Females have two X
chromosomes but males only have one. That means that for these 1300
genes on the X, females have two copies of each gene but males only
have one. We might speculate from this that female cells would
produce twice the amounts of proteins from these genes (referred to
as X-linked genes) as males.
But our knowledge of disorders like
Down’s syndrome makes this seem rather unlikely. Having three
copies of chromosome 21 (instead of the normal two) results in
Down’s syndrome, which is a major disorder in those individuals who
are born with the condition. Trisomies of most other chromosomes
are so severe that children are never born with these conditions,
because the embryos cannot develop properly. For example, no child
has ever been born who has three copies of chromosome 1 in all
their cells. If the 50 per cent increase in gene expression from an
autosome can cause such problems in trisomic conditions, how do we
explain the X chromosome scenario? How is it possible for females
to survive when they have twice as many X chromosome genes as
males? Or, to put it the other way – why are males viable if they
only have half as many X chromosome genes as females?
The answer is that expression of X-linked
genes is actually pretty much the same in males and females,
despite the different number of chromosomes, a phenomenon called
dosage compensation. The XY system of sex determination doesn’t
exist in other animal classes so X chromosome dosage compensation
is limited to placental mammals.
In the early 1960s a British geneticist
called Mary Lyon postulated how dosage compensation would occur at
the X chromosome. These were her predictions:
1. Cells from the normal female
would contain only one active X chromosome;
2. X inactivation would occur
early in development;
3. The inactive X could be
either maternally or paternally derived, and the inactivation would
be random in any one cell;
4. X inactivation would be
irreversible in a somatic cell and all its
descendants.
These predictions have proven
remarkably prescient4,5. So prescient, in fact,
that many textbooks refer to X inactivation as Lyonisation. We’ll
take the predictions one at a time:
1. Individual cells from a
normal female do indeed only express genes from one X chromosome
copy – the other copy is, effectively, shut down;
2. X inactivation occurs early
in development, at the stage when the pluripotent cells of the
embryonic inner cell mass are beginning to differentiate into
different lineages (near the top of Waddington’s epigenetic
landscape);
3. On average, in 50 per cent
of cells in a female the maternally derived X chromosome is shut
down. In the other 50 per cent of cells it’s the chromosome
inherited from Dad which gets inactivated;
4. Once a cell has switched off
one of a pair of X chromosomes, that particular copy of the X stays
switched off in all the daughter cells for the rest of that woman’s
life, even if she lives to over 100 years of age.
The X chromosome isn’t inactivated by
mutation; it keeps its DNA sequence entirely intact. X inactivation
is the epigenetic phenomenon par excellence.
X inactivation has proven to be a
remarkably fertile research field. Some of the mechanisms involved
have turned out to have parallels in a number of other epigenetic
and cellular processes. The consequences of X inactivation have
important implications for a number of human disorders and for
therapeutic cloning. Yet even now, 50 years on from Mary Lyon’s
ground-breaking work, there remain a number of mysteries about how
X inactivation actually takes place.
The more we ponder X inactivation, the
more extraordinary it appears. For a start, the inactivation is
only on the X chromosome, not on any of the autosomes, so the cell
must have a way of distinguishing X chromosomes and autosomes from
one another. Furthermore, the inactivation in the X doesn’t just
affect one or a few genes, such as occurs in imprinting. No, in X
inactivation, over 1,000 genes are turned off, for
decades.
Think of a car manufacturer, with a
factory in Japan and another in Germany. Imprinting is the
equivalent of a few changes in specification for the different
markets. The German factory may switch on the machine that installs
the heater on the steering wheel and switch off the robot that
inserts the automatic air freshener, whilst the Japanese factory
does the opposite. X inactivation is the equivalent of shutting
down and mothballing one factory, never to be re-opened unless the
company is bought by a new manufacturer.
Random inactivation
The other major difference between
X-inactivation and imprinting is that there is no parent-of-origin
effect in X imprinting. In somatic cells, it doesn’t matter if an X
chromosome was inherited from your mother or your father. Each has
a 50 per cent chance of being inactivated. The reason why this is
the case makes complete evolutionary sense.
Imprinting is about balancing out the
competing demands of the maternal and paternal genomes, especially
during development. The imprinting mechanisms that have evolved are
specifically targeted at individual genes, or small clusters of
genes, that particularly influence foetal growth. There are, after
all, only 50–100 imprinted genes in the mammalian
genome.
But X inactivation operates on a much
greater scale. It’s a mechanism for switching off over 1,000 genes,
en masse and permanently. A thousand genes is a lot, about 5 per
cent of the total number of protein-coding genes, so there’s always
a possibility that any given gene on an X chromosome may have a
mutation. Figure 9.2 compares the outcomes
of imprinted X inactivation on the left, with random X inactivation
on the right. For clarity, the diagram just exemplifies a mutation
in a paternally inherited gene, with imprinted inactivation of the
maternally derived X chromosome.
By using random X inactivation,
cells are able to minimise the effects of mutations in X-linked
genes.
It’s important to bear in mind that the
inactive X really is inactive. Almost all the genes are permanently
shut off and this inactivation cannot normally be broken. When we
refer to the active X chromosome, we are using slightly ambiguous
shorthand. It doesn’t mean that every gene on that X is active all
the time in every cell. Rather, the genes have the potential to be
active. They are subject to all the normal epigenetic modifications
and controls on gene expression, so that selected genes are
switched on or off in a controlled manner, in response to
developmental cues or environmental signals.
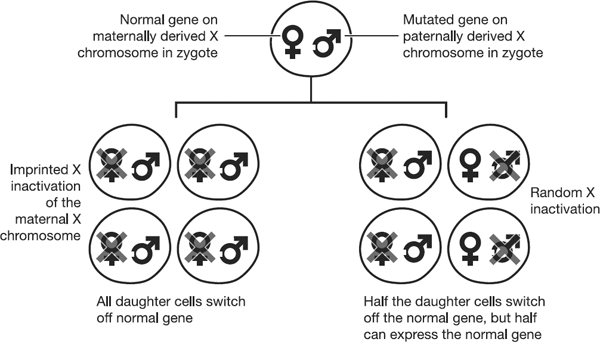
Figure 9.2 Each
circle represents a female cell, containing two X chromosomes. The
X chromosome inherited from the mother is indicated by the female
symbol. The X chromosome inherited from the father is indicated by
the male symbol, and contains a mutation, denoted by the white
square notch. The left hand side of the diagram demonstrates that
imprinted inactivation of the maternally derived X chromosome would
result in all cells of the body expressing only the X chromosome
carrying the mutation, which was inherited from the father. On the
right hand side, the X chromosomes are randomly inactivated,
independent of their parent-of-origin. As a result, on average,
half of the somatic cells will express the normal version of the X
chromosome. This makes random X inactivation a less risky
evolutionary strategy than imprinted X inactivation.
Women really are more complicated than
men
One interesting consequence of X
inactivation is that (epigenetically) females are more complicated
than males. Males only have one X chromosome in their cells, so
they don’t carry out X inactivation. But females randomly
inactivate an X chromosome in all their cells. Consequently, at a
very fundamental level, all cells in a female body can be split
into two camps depending on which X chromosome they inactivated.
The expression for this is that females are epigenetic
mosaics.
This sophisticated epigenetic control in
females is a complicated and highly regulated process, and that’s
where Mary Lyon’s predictions have provided such a useful
conceptual framework. They can be paraphrased as the following four
steps:
1. Counting: cells from the
normal female would contain only one active X
chromosome;
2. Choice: X inactivation would
occur early in development;
3. Initiation: the inactive X
could be either maternally or paternally derived, and the
inactivation would be random in any one cell;
4. Maintenance: X inactivation
would be irreversible in a somatic cell and all its
descendants.
Unravelling the mechanisms behind these
four processes has kept researchers busy for nearly 50 years, and
this effort is continuing today. The processes are incredibly
complicated and sometimes involve mechanisms that had barely been
imagined by any scientists. That’s not really surprising, because
Lyonisation is quite extraordinary – X inactivation is a procedure
where a cell treats two identical chromosomes in diametrically
opposite and mutually exclusive ways.
Experimentally, X inactivation is
challenging to investigate. It is a finely balanced system in
cells, and slight variations in technique may have a major impact
on the outcome of experiments. There’s also considerable debate
about the most appropriate species to study. Mouse cells have
traditionally been used as the experimental system of choice, but
we are now realising that mouse and human cells aren’t identical
with respect to X inactivation6. However, even allowing for
these ambiguities, a fascinating picture is beginning to
emerge.
Counting chromosomes
Mammalian cells must have a mechanism
to count how many X chromosomes they contain. This prevents the X
chromosome from being switched off in male cells. The importance of
this was shown in the 1980s by Davor Solter. He created embryos by
transferring male pronuclei into fertilised eggs. Males have an XY
karyotype, and when they produce gametes each individual sperm will
contain either an X or a Y. By taking pronuclei from different
sperm and injecting them into ‘empty’ eggs, it was possible to
create XX, XY or YY zygotes. None of these resulted in live births,
because a zygote requires both maternal and paternal inputs, as we
have already seen. But the results still told us something very
interesting, and are summarised in Figure
9.3.

Figure 9.3 Donor egg
reconstitution experiments were performed in which the donor egg
received a male and female pronucleus or two pronuclei from males.
Just as in Figure 7.2, the embryos
derived from two male pronuclei failed to develop to term. When the
nuclei each contained a Y chromosome, and no X chromosome, the
embryos failed at a very early stage. Embryos derived from two male
pronuclei where at least one contained an X chromosome developed
further before they also died.
The earliest loss of embryos occurred in
those that had been reconstituted from two male pronuclei which
each contained a Y chromosome as the sole sex chromosome7. In
these embryos there was no X chromosome at all, and this was
associated with exceptionally early developmental failure. This
shows that the X chromosome is clearly essential for viability.
This is why male (XY) cells need to be able to count, so that they
can recognise that they only contain one X, and thus avoid
inactivating it. Turning off the solitary X would be disastrous for
the cell.
Having counted the number of X
chromosomes, there must be a mechanism in female cells by which one
X is randomly selected for inactivation. Having selected a
chromosome, the cell starts the inactivation
procedure.
X inactivation happens early in female
embryogenesis, as the cells of the ICM begin to differentiate into
the different cell types of the body. Experimentally, it is
difficult to work on the small number of cells available from each
blastocyst so researchers typically use female ES cells. Both X
chromosomes are active in these cells, just like in the
undifferentiated ICM. It’s easy to roll ES cells down Waddington’s
epigenetic landscape, just by subtly altering the conditions in
which the cells are cultured in the lab. Once we change the
conditions to encourage the female ES cells to differentiate, they
begin to inactivate an X chromosome. Because ES cells can be grown
in almost limitless numbers in labs, this provides a convenient
model system for studying X inactivation.
Painting an X-rated picture
Initial insights into X inactivation
came from studying mice and cell lines with structurally rearranged
chromosomes. In some of these studies, various sections of an X
chromosome were missing. Depending on which parts were missing, the
X chromosome did or did not inactivate normally. In other studies,
sections had come off the X chromosome and attached themselves onto
an autosome. Depending on which part of the X chromosome had
transferred, this could result in switching off the structurally
abnormal autosome8,9.
These experiments showed that there was a
region on the X chromosome that was vitally important for X
inactivation. This region was dubbed the X Inactivation Centre. In
1991 a group from Hunt Willard’s lab at Stanford University in
California showed that the X Inactivation Centre contained a gene
that they called Xist, after X-inactive
(Xi) specific transcript10. This gene was only
expressed from the inactive X chromosome, not from the
active one. Because the gene was only expressed from one of the two
X chromosomes, this made it an attractive candidate as the
controller of X inactivation, where two identical chromosomes
behave non-identically.
Attempts were made to identify the
protein encoded by the Xist gene11
but by 1992 it was clear that there was something very strange
going on. The Xist gene was transcribed to form RNA copies.
The RNA was processed just like any other RNA. It was spliced, and
various structures were added to each end of the transcript to
improve its stability. So far, so normal. But before RNA molecules
can code for protein, they have to move out of the nucleus and into
the cytoplasm of the cell. This is because the ribosomes – the
intracellular factories that join amino acids into long protein
chains – are only found in the cytoplasm. But the Xist RNA
never moved out of the nucleus, which meant it could never generate
a protein12,13.
This at least cleared up one thing that
had puzzled the scientific community when the Xist gene was
first identified. Mature Xist RNA is a long molecule, of
about 17,000 base-pairs (17kb). One amino acid is coded for by a
three base-pair codon, as described in Chapter
3. Therefore, in theory, the 17,000 base-pairs of Xist
should be able to code for a protein of about 5,700 amino acids.
But when researchers analysed the Xist sequence with protein
prediction programs, they simply couldn’t see how it could encode
anything this long. There were stop codons (which signal the end of
a protein) all through the Xist sequence and the longest
predicted run without stop codons was only enough to code for 298
amino acids (894 base-pairs14). Why would a gene have
evolved which created a 17kb transcript, but only used about 5 per
cent of this to encode protein? That would be a very inefficient
use of energy and resources in a cell.
But since Xist never actually
leaves the nucleus, its lack of potential protein coding is
irrelevant. Xist doesn’t act as a messenger RNA (mRNA) that
transmits the code for a protein. It is a class of molecule called
a non-coding RNA (ncRNA). Xist may not code for protein, but
this doesn’t mean it has no activity. Instead, the Xist
ncRNA itself acts as a functional molecule, and it is critical for
X inactivation.
Back in 1992 ncRNAs were a real novelty,
and only one other was known at the time. Even now, there is
something very unusual about Xist. It’s not just that it
doesn’t leave the nucleus. Xist doesn’t even leave the
chromosome that produces it. When ES cells begin to differentiate,
only one of the chromosomes produces Xist RNA. This is the
chromosome that will be the inactive one. Xist doesn’t move
away from the chromosome that produced it. Instead, it binds to the
chromosome and starts to spread out along it.
Xist is often described as
‘painting’ the inactive X and it’s a very good description. Let’s
revert yet again to our analogy of the DNA code as a script. This
time we’ll imagine that the script is written on a wall, maybe it’s
an inspiring poem or speech in a classroom. At the end of the
summer term the school building closes down and is sold for
conversion to apartments. The decorators arrive and paint over the
script. Now there’s nothing to tell the new residents to ‘play up
and play the game’, or exactly how they should ‘meet with Triumph
and Disaster’. But the instructions are actually still there,
they’re just hidden from view.
When Xist binds over the X
chromosome that produced it, it induces a kind of creeping
epigenetic paralysis. It covers more and more genes, switching them
off. It first seems to do this by acting as a barrier between the
genes and the enzymes that normally copy them into mRNA. But as the
X inactivation gets better established, it changes the epigenetic
modifications on the chromosome. The histone modifications that
normally turn genes on are removed. They are replaced by repressive
histone modifications that turn genes off.
Some of the normal histones are removed
altogether. Histone H2A is replaced by a related but subtly
different molecule called macroH2A, strongly associated with gene
repression. The promoters of genes undergo DNA methylation, an even
more stringent way of turning the genes off. All these changes lead
to binding of more and more repressor molecules, coating the DNA on
the inactive X and making it less and less accessible to the
enzymes that transcribe genes. Eventually, the DNA on the X
chromosome gets incredibly tightly wound up, like a giant wet towel
being turned at each end, and the whole chromosome moves to the
edge of the nucleus. By this stage most of the X chromosome is
completely inactive, except for the Xist gene, which is a
little pool of activity in the midst of a transcriptional
desert15.
Whenever a cell divides, the
modifications to the inactive X are copied over from mother cell to
daughter cell, and so the same X remains inactivated in all
subsequent generations of that starter cell.
While the effects of Xist are
amazing, the description above still leaves a lot of questions
unanswered. How is Xist expression controlled? Why does it
switch on when ES cells start to differentiate? Is Xist only
functional when it’s in female cells, or could it act in males
cells too?
The power of a kiss
The last question was first addressed
in the lab of Rudi Jaenisch, whom we met in the context of iPS
cells and Shinya Yamanaka’s work in Chapter
2. In 1996, Professor Jaenisch and his colleagues created mice
carrying a genetically engineered version of the X Inactivation
Centre (an X Inactivation Centre transgene). This was 450kb in
size, and included the Xist gene plus other sequences on
either side. They inserted this into an autosome (non-sex
chromosome), created male mice carrying this transgene, and studied
ES cells from these mice. The male mice only contained one normal X
chromosome, because they have the XY karyotype. However, they had
two X Inactivation Centres. One was on the normal X chromosome, and
one was on the transgene on the autosome. When the researchers
differentiated the ES cells from these mice, they found that
Xist could be expressed from either of the X Inactivation
Centres. When Xist was expressed, it inactivated the
chromosome from which it was expressed, even if this was the
autosome carrying the transgene16.
These experiments showed that even cells
that are normally male (XY) can count their X chromosomes.
Actually, to be more specific, it showed they could count their X
Inactivation Centres. The data also demonstrated that the critical
features for counting, choosing and initiation were all present in
the 450kb of the X Inactivation Centre around the Xist
gene.
We know a bit more now about the
mechanism of chromosome counting. Cells don’t normally count their
autosomes. Both copies of chromosome 1, for example, operate
independently. But we know that the two copies of the X chromosome
in a female ES cell somehow communicate with each other. When X
inactivation is getting going, the two X chromosomes in a cell do
something very weird.
They kiss.
That’s a very anthropomorphic way of
describing the event, but it’s a pretty good description. The
‘kiss’ only lasts a couple of hours or so, and it’s startling to
think this sets a pattern that can persist in cells for the next
hundred years, if a woman lives that long. This chromosomal smooch
was first shown in 1996 by Jeannie Lee, who started out as a
post-doctoral researcher in Rudi Jaenisch’s lab, but who is now a
professor in her own right at Harvard Medical School, where she was
one of the youngest professors ever appointed. She showed that
essentially the two copies of the X find each other and make
physical contact. This physical contact is only over a really small
fraction of the whole chromosome, but it’s essential for triggering
inactivation17. If it doesn’t happen, then
the X chromosome assumes it is all alone in the cell, Xist
never gets switched on, and there is no X inactivation. This is a
key stage in chromosome counting.
It was Jeannie Lee’s lab that also
identified one of the critical genes that controls Xist
expression18. DNA is double-stranded,
with the bases in the middle holding the strands together. Although
we often envisage it as looking like a railway track, it might be
better to think of it as two cable cars, running in opposite
directions. If we use this metaphor, then the X Inactivation Centre
looks a bit like Figure 9.4.
There is another non-coding RNA, about
40kb in length, in the same stretch of DNA as Xist. It
overlaps with Xist but is on the opposite strand of the DNA
molecule. It is transcribed into RNA in the opposite direction to
Xist and is referred to as an antisense transcript. Its name
is Tsix. The eagle-eyed reader will notice that Tsix
is Xist backwards, which has an unexpectedly elegant logic
to it.
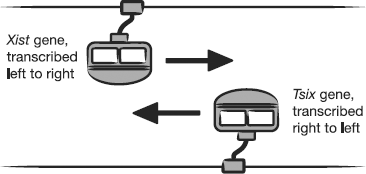
Figure 9.4 The two
strands of DNA at a specific location on the X chromosome can each
be copied to create mRNA molecules. The two backbones are copied in
opposite directions to each other, allowing the same region of the
X chromosome to produce Xist RNA or Tsix
RNA.
This overlap in location between
Tsix and Xist is really significant in terms of how
they interact, but it makes it exceedingly tricky to perform
conclusive experiments. That’s because it’s very difficult to
mutate one of the genes without mutating its partner on the
opposite strand, a sort of collateral damage. Despite this,
considerable strides have been made in understanding how
Tsix influences Xist.
If an X chromosome expresses Tsix,
this prevents Xist expression from the same chromosome.
Oddly enough, it may be the simple action of transcribing
Tsix that prevents the Xist expression, rather than
the Tsix ncRNA itself. This is analogous to a mortice lock.
If I lock a mortice from the inside of my house and leave the key
in the lock, my partner can’t unlock the door from the outside of
the house. I don’t need to keep locking the door, just having the
key in there is enough to stop the action of someone on the other
side. So, when Tsix is switched on, Xist is switched
off and the X chromosome is active.
This is the situation in ES cells, where
both X chromosomes are active. Once the ES cells begin to
differentiate, one of the pair stops expressing Tsix. This
allows expression of Xist from that X chromosome, which
drives X inactivation.
Tsix alone is probably not enough
to keep Xist repressed. In ES cells, the proteins Oct4, Sox2
and Nanog bind to the first intron of Xist and suppress its
expression19. Oct4 and Sox2 were two of
the four factors used by Shinya Yamanaka when he reprogrammed
somatic cells to the pluripotent iPS cell type. Subsequent
experiments showed that Nanog (named after the mythical Celtic land
of everlasting youth) can also work as a reprogramming factor.
Oct4, Sox2 and Nanog are highly expressed in undifferentiated cells
like ES cells, but their levels fall as cells start to
differentiate. When this happens in differentiating female ES
cells, Oct4, Sox2 and Nanog stop binding to the Xist intron.
This removes some of the barriers to Xist expression.
Conversely, when female somatic cells are reprogrammed using the
Yamanaka approach, the inactive X chromosome is reactivated20.
The only other time the inactive X is reactivated is during the
formation of primordial germ cells in development, which is why the
zygote starts out with two active X chromosomes.
We are still a bit vague as to why X
inactivation is so mutually exclusive between the pair of
chromosomes. One theory is that it’s all down to what happens when
the X chromosomes kiss. This happens at a developmental point where
Tsix levels are starting to fall, and the levels of the
Yamanaka factors are also declining. The theory is that the pair of
chromosomes reaches some sort of compromise. Rather than each
ending up with a sub-optimal amount of non-coding RNAs and other
factors, the binding molecules all get shunted together onto one of
the pair. There’s not a great deal of clarity on how this happens.
It could be that one of the pair of chromosomes just by chance
carries slightly more of a key factor than the other. This makes it
slightly more attractive to certain proteins. Complexes may build
up in a self-sustaining way, so that the more of a complex one
chromosome starts with, the more it can drag off its partner. The
rich get richer, the poor get poorer …
It’s quite remarkable how many gaps
remain in our understanding of X inactivation, 50 years after Mary
Lyon’s formative work. We don’t even really understand how the
Xist RNA ends up coating the chromosome from which it is
expressed, or how it recruits all those negative repressive
epigenetic enzymes and modifications. So perhaps it’s timely to
move off the shifting sands and step back onto more solid
ground.
Let’s return to this statement from
earlier in the chapter: ‘Once a cell has switched off one of a pair
of X chromosomes, that particular copy of the X stays switched off
in all the daughter cells for the rest of that woman’s life, even
if she lives to over a hundred years of age.’ How do we know that?
How can we be so certain that X inactivation is stable in somatic
cells? It is now possible to perform genetic manipulation to show
this in species like mice. But long before that became feasible
scientists were already pretty certain this was the case. For this
piece of information we thank not mice, but cats.
Learning from the epigenetic
cat
Not just any old cats, but specifically
tortoiseshell ones. You probably know how to recognise a classic
tortoiseshell cat. It’s the one that’s a mixture of black and
ginger splodges, sometimes on a white background. The colour of
each hair in a cat’s coat is caused by cells called melanocytes
that produce pigment. Melanocytes are found in the skin, and
develop from special stem cells. When melanocyte stem cells divide,
the daughter cells stay close to each other, forming a little patch
of clonal cells from the same parent stem cell.
Now, here’s an amazing thing: if a cat’s
colour is tortoiseshell, it’s a female.
There is a gene for coat colour that
encodes either black pigment or orange pigment. This gene is
carried on the X chromosome. A cat may receive the black version of
the gene on the X chromosome inherited from her mother and the
orange version on the X chromosome inherited from her father (or
vice versa). Figure 9.5 shows what
happens next.
So the tortoiseshell cat ends up with
patches of orange and patches of black, depending on the X
chromosome that was randomly inactivated in the melanocyte stem
cell. The pattern won’t change as the cat gets older, it stays the
same throughout its life. That tells us that the X inactivation
stays the same in the cells that create this coat
pattern.
We know that tortoiseshell cats are
always female because the gene for the coat colour is only on the X
chromosome, not the Y. A male cat only has one X chromosome, so it
could have black fur or ginger fur, but never both.

Figure 9.5 In female
tortoiseshell cats, the genes for orange and black fur are carried
on the X chromosome. Depending on the pattern of X chromosome
inactivation in the skin, clonal patches of cells will give rise to
discrete patterns of orange and black fur.
Something rather similar happens in a
rare human disorder called X-linked hypohidrotic ectodermal
dysplasia. This condition is caused by mutations in a gene called
ECTODYSPLASIN-A, carried on the X chromosome21. A
male with a mutation in his sole copy of ECTODYSPLASIN-A on
his single X chromosome has a variety of symptoms, including a
total lack of sweat glands. This might sound socially advantageous,
but is actually incredibly dangerous. Sweating is one of the major
routes by which we lose excess heat, and men with this condition
are at serious risk of tissue damage or even death as a result of
heat stroke22.
Females have two copies of the
ECTODYSPLASIN-A gene, one on each of their X chromosomes. In
female carriers of X-linked hypohidrotic ectodermal dysplasia, one
X carries a normal copy of the gene, and one a mutated version.
There will be random inactivation of one X chromosome in different
cells. This means some cells will express a normal copy of
ECTODYSPLASIN-A. Other cells will randomly shut down the X
carrying the normal copy of the gene, and won’t be able to express
the ECTODYSPLASIN-A protein. Because of the clonal way in which
areas of skin develop, just like in the tortoiseshell cat, these
women have some patches of skin that express ECTODYSPLASIN-A and
some that don’t. Where there’s no ECTODYSPLASIN-A, the skin can’t
form sweat glands. As a consequence, these women have patches of
skin that can sweat and cool down, and others that
can’t.
Random X inactivation can significantly
influence how females are affected by mutations in genes on the X
chromosome. This depends not just on the type of gene that is
mutated but also on the tissues that express and require the
protein encoded by that gene. The disease called
mucopolysaccharidosis II (MPSII) is caused by mutations in the
LYSOSOMAL IDURONATE-2-SULFATASE gene, on the X chromosome.
Boys with this mutation on their single X chromosome are unable to
break down certain large molecules and these build up to toxic
levels in cells. The main symptoms include airway infections, short
stature and enlargement of the spleen and liver. Severely affected
boys also suffer mental retardation, and may die in their teenage
years.
Females with a mutation in the same gene
are usually perfectly healthy. LYSOSOMAL IDURONATE-2-SULFATASE
protein is usually secreted out of the cell that makes it and taken
up by neighbouring cells. In this situation it doesn’t matter too
much which X chromosome has been mutated in a specific cell. For
every cell that has inactivated the X carrying the normal version
of the gene, there is likely to be another cell nearby which
inactivated the other X chromosome and is secreting the protein.
This way, all cells end up with sufficient LYSOSOMAL
IDURONATE-2-SULFATASE protein, whether they produce it themselves
or not23.
Duchenne muscular dystrophy is a severe
muscle wasting disease caused by mutations in the X-linked
DYSTROPHIN gene. This is a large gene that encodes a big
protein which acts as an essential shock absorber in muscle fibres.
Boys carrying certain mutations in DYSTROPHIN suffer major
muscle loss that usually results in death in the teenage years.
Females with the same mutation are usually symptom-free. The reason
for this is that muscle has a very unusual structure. It is called
a syncytial tissue, which means that lots of individual cells fuse
and operate almost like one giant cell, but with lots of discrete
nuclei. This is why most females with a DYSTROPHIN mutation
are symptom-free. There is enough normal DYSTROPHIN protein encoded
by the nuclei that switched off the mutated DYSTROPHIN gene
to keep this syncytial tissue functioning healthily24.
There are occasional cases where this
system breaks down. There was a case of female monozygotic twins
where one twin was severely affected by Duchenne muscular dystrophy
and the other was healthy25. In the affected twin, the
X inactivation had become skewed. Early in tissue differentiation
the majority of her cells that would give rise to muscle happened,
by ill chance, to switch off the X chromosome carrying the normal
copy of the DYSTROPHIN gene. Thus, most of the muscle tissue
in this woman only expressed the mutated version of DYSTROPHIN, and
she developed severe muscle wasting. This could be considered the
ultimate demonstration of the power of a random epigenetic event.
Two identical individuals, each with two apparently identical X
chromosomes, had a completely discordant phenotype, because of a
shift in the epigenetic balance of power.
Sometimes, however, it is essential that
individual cells express the correct amount of a protein.
You may have noticed in Chapter 4 that Rett
syndrome only affected girls. One might hypothesise that boys are
somehow very resistant to the effects of the MeCP2 mutation,
but actually the opposite is true. MeCP2 is carried on the X
chromosome so a male foetus that inherits a Rett syndrome mutation
in this gene has no means of expressing normal MeCP2 protein. A
complete lack of normal MeCP2 expression is generally lethal in
early development, and that’s why very few boys are born with Rett
syndrome. Girls have two copies of the MeCP2 gene, one on
each X chromosome. In any given cell, there is a 50 per cent chance
that the cell will inactivate the X that carries the unmutated
MeCP2 gene and that the cell will not express normal MeCP2
protein. Although a female foetus can develop, there are ultimately
major effects on normal post-natal brain development and function
when a substantial number of neurons lack MeCP2
protein.
One, two, many
There are other issues that can develop
around the X chromosome. One of the questions we need to answer
about X inactivation, is how good mammalian cells are at counting.
In 2004 Peter Gordon of Columbia University in New York reported on
his studies on the Piraha tribe in an isolated region of Brazil.
This tribe had numbers for one and two. Everything beyond two was
described by a word roughly equating to ‘many’26.
Are our cells the same, or can they count above two? If a nucleus
contains more than two X chromosomes, can the X inactivation
machinery recognise this, and deal with the consequences? Various
studies have shown that it can. Essentially, no matter how many X
chromosomes (or more strictly speaking X Inactivation Centres) are
present in a nucleus, the cell can count them and then inactivate
multiple X chromosomes until there is only one remaining
active.
This is the reason why abnormal numbers
of X chromosomes are relatively frequent in humans, in contrast to
abnormalities in the number of autosomes. The commonest examples
are shown in Table 9.1.
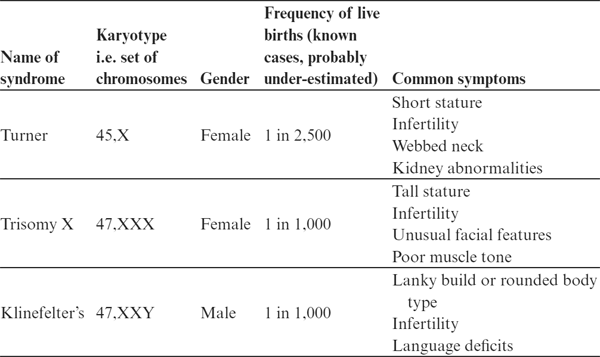
Table 9.1 Summary of
the major characteristics of the commonest abnormalities in sex
chromosome number in humans.
The infertility that is a feature of all
these disorders is in part due to problems when creating eggs or
sperm, where it’s important that chromosomes line up in their
pairs. If there is an uneven total number of sex chromosomes this
stage goes wrong and formation of gametes is severely
compromised.
Leaving aside the infertility, there are
two obvious conclusions we can draw from this table. The first is
that the phenotypes are all relatively mild compared with, for
example, trisomy of chromosome 21 (Down’s syndrome). This suggests
that cells can tolerate having too many or too few copies of the X
chromosome much better than having extra copies of an autosome. But
the other obvious conclusion is that an abnormal number of X
chromosomes does indeed have some effects on
phenotype.
Why should this be? After all, X
inactivation ensures that no matter how many X chromosomes are
present, all bar one get inactivated early in development. But if
this was the end of the story there would be no difference in
phenotype between 45, X females compared with 47, XXX females or
with the normal 46, XX female constitution. Similarly, males with
the normal 46, XY karyotype should be phenotypically identical to
males with the 47, XXY karyotype. In all of these cases there
should be only one active X chromosome in the cells.
One thought as to why people with these
karyotypes were clinically different was that maybe X inactivation
is a bit inefficient in some cells, but this doesn’t seem to be the
case. X inactivation is established very early in development and
is the most stable of all epigenetic processes. An alternative
explanation was required.
The answer has its origin about 150
million years ago, when the XY system of sex determination in
placental mammals first developed. The X and Y chromosomes are
probably descendants of autosomes. The Y chromosome has changed
dramatically, the X chromosome much less so27.
However, both retain shadows of their autosomal past. There are
regions on both the X and the Y called pseudoautosomal regions. The
genes in these regions are found on both the X and the Y
chromosome, just in the same way as pairs of autosomes have the
same genes in the same positions, one inherited from each
parent.
When an X chromosome inactivates, these
pseudoautosomal regions are spared. This means that, unlike most
X-linked genes, those in the pseudoautosomal regions don’t get
switched off. Consequently, normal cells potentially express two
copies of these genes in all cells. The two copies are expressed
either from the two X chromosomes in a normal female or from the X
and the Y in a normal male.
But in Turner’s syndrome, the affected
female only has one X chromosome, so she expresses only one copy of
the genes in the pseudoautosomal region, half as much as normal. In
Trisomy X, on the other hand, there are three copies of the genes
in the pseudoautosomal regions. As a result, the cells in an
affected region will produce proteins from these genes at 50 per
cent above the normal level.
One of the genes in the X chromosome
pseudoautosomal regions is called SHOX. Patients with
mutations in this gene have short stature. It is likely that this
is also why patients with Turner’s syndrome tend to be short – they
don’t produce enough SHOX protein in their cells. By contrast,
patients with Trisomy X are likely to produce 50 per cent more SHOX
protein than normal, which is probably why they tend to be
tall28.
It’s not just humans who have trisomies
of the sex chromosomes. One day you may be happily amazing your
friends with your confident statement that their tortoiseshell cat
is female when they deflate you by telling you that their pet has
been sexed by the vet and is actually a Tom. At this point, smile
smugly and then say ‘Oh, in that case he’s karyotypically abnormal.
He has an XXY karyotype, rather than XY’. And if you’re feeling
particularly mean, you can tell them that Tom is infertile. That
should shut them up.